Elastic Constants
Solids respond to small external loads through a reversible elastic response. The strength of the response is characterized by the elastic moduli. matscipy.elasticity implements functions for computing elastic moduli from small deformation of atomistic systems that consider potential symmetries of the underlying atomic system, in particular for crystals. matscipy.elasticity also implements analytic calculation of elastic moduli for some interatomic potentials, described in more detail below. The computation of elastic moduli is a basic prerequisite for multi-scale modelling of materials, as they are the most basic parameters of continuum material models.
In this tutorial, we will go over different ways that matscipy can compute elastic constants of an atomic configuration.
Problem setup
Let’s first create an FCC system and view it interactively:
from ase.lattice.cubic import FaceCenteredCubic
def interactive_view(system):
from ase.visualize import view
# Interactive view of the lattice
v = view(system, viewer='ngl')
# Resize widget
v.view._remote_call("setSize", target="Widget", args=["300px", "300px"])
v.view.center()
return v
# Define FCC crystal
system = FaceCenteredCubic(directions=[[1, 0, 0], [0, 1, 0], [0, 0, 1]], size=(3,3,3), symbol='Cu', pbc=(1,1,1))
interactive_view(system)
We’ll assign a force-field to this system based on the Lennard-Jones potential (LennardJonesCut) implemented in matscipy.
from matscipy.calculators.pair_potential import PairPotential, LennardJonesCut
from ase.data import reference_states, atomic_numbers
import numpy as np
Cu_num = atomic_numbers["Cu"]
lattice = reference_states[Cu_num]["a"]
sigma = lattice / (2**(2/3))
system.calc = PairPotential({
# Type map: define Copper-Copper pair interaction
(Cu_num, Cu_num): LennardJonesCut(epsilon=1, sigma=lattice * 2**(-1/6) / np.sqrt(2), cutoff=3 * lattice)
})
We test we have a sensible potential energy and that we have equilibrium.
from numpy.testing import assert_allclose
from IPython.display import display, Latex
display(Latex(f"$E_\\mathrm{{pot}} = {system.get_potential_energy():.1f}$"))
# Testing negative potential energy
assert system.get_potential_energy() < 0
# Testing equilibrium
assert_allclose(system.get_forces(), 0, atol=3e-14)
Crystalline elastic constants
Let us first compute elastic constants with the matscipy.elasticity module, with two different functions:
measure_triclinic_elastic_constantscomputes the full Hooke’s tensor by finite differencesfit_elastic_constantscomputes a range of deformed configurations and fits a Hooke’s tensor with least-squares
from matscipy.elasticity import measure_triclinic_elastic_constants, fit_elastic_constants
from matscipy.elasticity import full_3x3x3x3_to_Voigt_6x6
C_finite_differences = full_3x3x3x3_to_Voigt_6x6(measure_triclinic_elastic_constants(system))
C_least_squares, _ = fit_elastic_constants(system, verbose=False)
Let’s plot the Hooke tensor:
import matplotlib.pyplot as plt
def spy_constants(ax, constants):
ax.imshow(constants, cmap='RdPu', interpolation='none')
labels = np.full_like(constants, "", dtype=object)
labels[:3, :3] = "$\\lambda$\n"
labels[(np.arange(3), np.arange(3))] = "$\\lambda + 2\\mu$\n"
labels[(np.arange(3, 6), np.arange(3, 6))] = "$\\mu$\n"
max_C = constants.max()
for i in range(constants.shape[0]):
for j in range(constants.shape[1]):
color = "white" if constants[i, j] / max_C > 0.7 else "black"
numeric = f"${constants[i, j]:.2f}$" if np.abs(constants[i, j]) / max_C > 1e-3 else "$\\sim 0$"
ax.annotate(labels[i, j] + numeric,
(i, j),
horizontalalignment='center',
verticalalignment='center', color=color)
ax.set_xticks(np.arange(constants.shape[1]))
ax.set_yticks(np.arange(constants.shape[0]))
ax.set_xticklabels([f"C$_{{i{j+1}}}$" for j in range(constants.shape[1])])
ax.set_yticklabels([f"C$_{{{i+1}j}}$" for i in range(constants.shape[0])])
fig, axs = plt.subplots(1, 2, figsize=(9, 5))
spy_constants(axs[0], C_finite_differences)
spy_constants(axs[1], C_least_squares)
axs[0].set_title(f"Finite differences")
axs[1].set_title(f"Least squares")
plt.show()
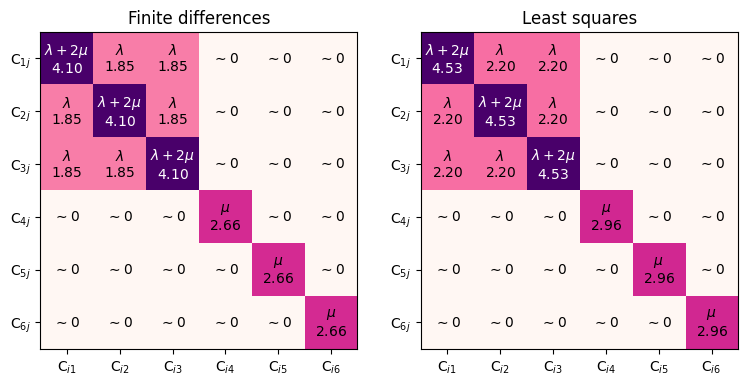
With second-order derivatives
Most calculators in matscipy actually implement second-order derivatives, and can therefore directly compute elastic constants:
C = full_3x3x3x3_to_Voigt_6x6(system.calc.get_property("elastic_constants", system))
fig, axs = plt.subplots(1, 2, figsize=(9, 5))
spy_constants(axs[0], C_least_squares)
spy_constants(axs[1], C)
axs[0].set_title(f"Least squares")
axs[1].set_title(f"Direct evaluation")
plt.show()
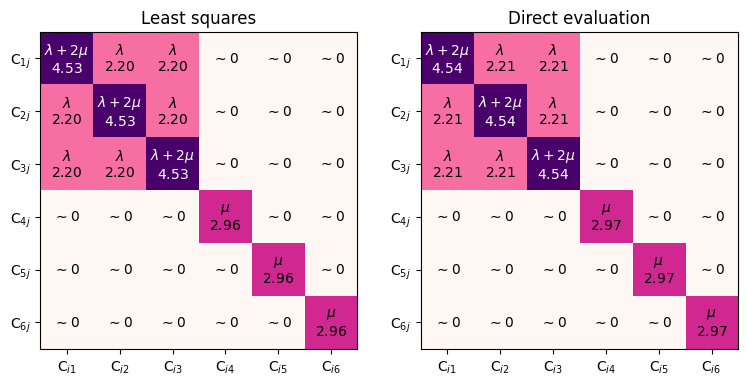
Amorphous elastic constants
In amorphous solids, non-affine relaxation modes play an important role in elastic deformation.
Problem setup
Let’s randomize our atoms to mimic a glassy structure:
from ase.io import read
from pathlib import Path
data_path = Path("..") / ".." / "tests"
system = read(data_path / "CuZr_glass_460_atoms.gz")
interactive_view(system)
Setting up the calculator:
from matscipy.calculators.eam import EAM
system.calc = EAM(data_path / 'ZrCu.onecolumn.eam.alloy')
display(Latex(f"$E_\\mathrm{{pot}} = {system.get_potential_energy():.1f}$"))
Fitting elastic constants
The fit_elastic_constants function accepts a minimizer procedure as argument to account for non-affine relaxation modes.
from ase.optimize import FIRE
delta = 1e-4 # Configuration change increment
C_affine, _ = fit_elastic_constants(system, verbose=False, delta=delta)
C_relaxed, _ = fit_elastic_constants(system, verbose=False, delta=delta,
# adjust fmax to desired precision
optimizer=FIRE, fmax=5 * delta)
fig, axs = plt.subplots(1, 2, figsize=(9, 5))
spy_constants(axs[0], C_affine)
spy_constants(axs[1], C_relaxed)
axs[0].set_title(f"Affine only")
axs[1].set_title(f"Affine + Non-affine")
plt.show()
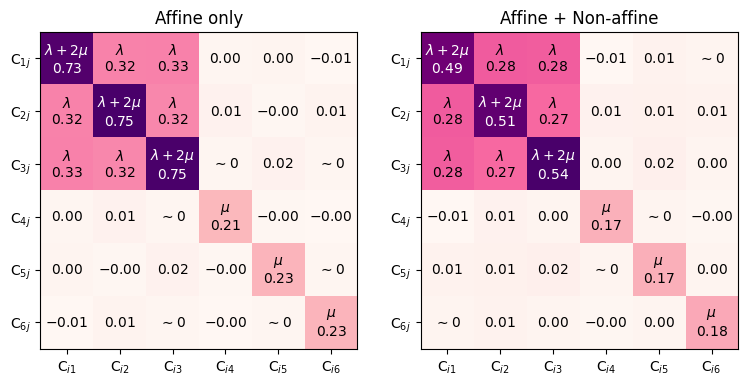
One can see that elastic constants are significantly reduced when the internal relaxation is included. However, mind that the reduction is very dependent on the optimizer’s stopping criterion fmax, which should ideally be lower than the deformation increment (we set it higher in the example above for demonstration purposes only).