Getting started¶
List of input parameters¶
- ns_run.usage()[source]¶
Print help to the standard output about the usage of the code and input parameters. The current list of parameters is the following:
max_volume_per_atom=float¶- Maximum volume per atom allowed during the run.default: 1.0e3
min_volume_per_atom=float¶- Minimum volume per atom allowed during the run.default: 1.0
start_species=int int [ float ] [, int int [ float ] ... ]¶- MANDATORY (or start_config_file)Atomic number; multiplicity; [ not recommended: mass (amu) ]. Info repeated for each species, separated by commas, mass is optional and not recommended.
start_config_file=str¶- MANDATORY (or start_species)Name of file to read in for atom informationdefault: ‘’
- ``keep_atoms_fixed=int ``¶
- Number of atoms to fix in surface simulation.
- ``apply_Z_wall=[T|F] ``¶
- Whether to have a boundary in the Z direction to keep free particles reaching the other side of “surface” layer due to periodic boundary conditions.This functionality is not fully tested! Recommended use with MC evaluator and fortran. If constructing a surface layer, make it parallel to the XY plane and set the X dimension with the “wall” taken into account, no atoms should violate the wall restriction initially!!!If True, the wall is set at 10 angstroms below the top of the simulation cell.
restart_file=path_to_file¶- File for restart configs. Mutually exclusive with
start_*, one is required. The file should contain the state of the walkers to continue from along with the restart iteration number. Normally such a file can be the concatenated snapshot files. n_walkers=int¶- MANDATORYTotal number of walkers, i.e. the size of the live set used. It has to be the multiple of the number of processors used and it also has to be larger than the number of processors.
n_cull=int¶- Number of walkers to kill at each NS iteration. Use of default 1 is strongly recommended.deafult: 1
n_extra_walk_per_task=int¶- default: 0
n_iter_times_fraction_killed=int¶- MANDATORYNumber of nested sampling iteration cycles performed per walker. Thus the total number of iterations will be
n_iter_times_fraction_killed/(n_cull/n_walkers). Either this orconverge_down_to_Tis required. converge_down_to_T=flot¶- MANDATORYtemperature down to which Z(T) should be converged. Either this or
n_iter_times_fraction_killedis required. T_estimate_finite_diff_lag=int¶- default: 1000Lag (in iterations) for doing d log Omega / d E finite difference derivative
min_Emax=float¶- Termination condition based on Emax: if this value is reached, the iteration will stop.No default.
out_file_prefix=str¶- String used as prefix for the different output files.No default.
energy_calculator= ( ASE | lammps | internal | fortran)¶- Energy calculator.default: fortran
n_extra_data=int¶- Amount of extra data per atom to pass around.default: 0
KEmax_max_T=float¶- If > 0, maximum temperature for estimating KEmax. For constant P ensemble 3/2 P Vmax will be used if KEmax_max_T is < 0.default: -1
kB=float¶- Boltzmann constantdefault: 8.6173324e-5 (eV/A)
start_energy_ceiling_per_atom=float¶- Maximum potential energy per atom for initial configurations. P*Vmax is added to this automatically in case of NpT runs.default: 1.0e9
start_energy_ceiling=float¶- DEPRECATED: use
start_energy_ceiling_per_atom. Maximum potential energy for initial configurations. P*Vmax is added to this automatically in case of NpT runs.default: 1.0e9 random_init_max_n_tries=int¶- Maximum number of tries to create initial random atomic configurationdefault 100
n_model_calls_expected=int¶- Total number of model calls performed during the random walk, expected by the user. The actual number of model calls performed during runtime can be different: this number is divided up among the processors in case of running parallel, and in order to make sure different processors perform about the same number of calls, it can be increased for some. (It is recommended to use n_model_calls_expected/[number of processors] > 20) Either this or the keyword
n_model_callsis mandatory, but the use of this keyword is strongly recommended. If < 0, value will be set to _sum_ of each type of steps (accounting for atom_traj_len), son_*_stepswill be setting the _number_ of calls, rather than just the ratios.default: 0 n_model_calls=int¶- Number of model calls performed during the random walk. This is the actual number of calls performed, ideally the program sets its value depending on n_model_calls_expected and the number of processors used. Either this or the keyword
n_model_calls_expectedis mandatory, though the usage of the latter is strongly recommended.default: 0 do_good_load_balance=[T | F]¶- Whether to do steps in groups with good load balancedefault: F
n_atom_steps=int¶- Ratio of atomic trajectories will be determined by
n_atom_steps/SUM(n_*_steps).default: 1 atom_traj_len=int¶- Length of atomic trajectory (MD steps or MC sweeps) in each atomic type step.default: 8
atom_traj_len_cost_multiplier=int¶- Multiplier for cost of an atomic step (set to 1 for MD, TE-HMC, and SP-MC with O(1) cost moves, N for naive SP-MC)default: 1
break_up_atom_traj=[T | F]¶- Whether to intersperse
n_atom_stepsatomic sub-trajectories with other types of steps.default: F n_cell_volume_steps=int¶- Ratio of cell volume steps will be determined by
n_cell_volume_steps/SUM(n_*_steps).default: 1 n_cell_shear_steps=int¶- Ratio of cell shear steps will be determined by
n_cell_shear_steps/SUM(n_*_steps).default: 1 n_cell_stretch_steps=int¶- Ratio of cell stretch steps will be determined by
n_cell_stretch_steps/SUM(n_*_steps).default: 1 n_swap_steps=int¶- Ratio of species swap steps will be determined by
n_swap_steps/SUM(n_*_steps). It has to be set other than zero for a multicomponent system.default: 0 swap_max_cluster=int¶- Maximum size of interconnected cluster to try to swap.default: 1
swap_r_cut=float¶- Cutoff radius for defining connected atoms for cluster.default: 2.5
swap_cluster_probability_increment=float¶- Factor between prob. of picking increasing larger clusters.default: 0.75
swap_velo=[T | F]¶- If true, swap velocities when swapping atoms, breaking coherence a bit.default: F
no_swap_velo_fix_mag_alt=[T | F]¶- If true, use alternate method for correcting velocity magnitudes when not swapping velocities.default: F
n_semi_grand_steps=int¶- Ratio of species type-change steps will be determined by
n_semi_grand_steps/SUM(n_*_steps). Only makes sense for a multicomponent system.default: 0 - ``semi_grand_potentials=Z1¶
- Chemical potentials for semi-grand canonical species-type transmutationsdefault: ‘’
velo_traj_len=int¶- Number of MC steps in (optional) explicit velocity MC trajectory.default: 0
random_energy_perturbation=float¶- default: 1.0e-12
atom_algorithm=[MC | MD | GMC]¶- MANDATORYUse either Monte Carlo or Molecular dynamics to explore. GMC is an alias for atom_algorithm=MC, MC_atom_Galilean=True.
MC_atom_velocities=[T | F]¶- This keyword is supported only for energy_calculator=fortran.default: F
MC_atom_velocities_pre_perturb=[T | F]¶- Perturb velocities (rejection free) before MC + velocities walk.default: F
MC_atom_step_size=float¶- Initial atom move step size in units of (max_volume_per_atom * N_atoms)^(1/3)default: 1.0
MC_atom_step_size_max=float¶- Maximum atom step size in units of (max_volume_per_atom * N_atoms)^(1/3).default: 1.0
MC_atom_uniform_rv=[T | F]¶- default: F
MC_atom_Galilean=[T | F]¶- default: F
GMC_no_reverse=[T | F]¶- default: T
MD_atom_velo_pre_perturb=[T | F]¶- Perturb velocities before MD trajectorydefault: F
MD_atom_velo_post_perturb=[T | F]¶- Perturb velocities after MD trajectorydefault: T
MD_atom_velo_flip_accept=[T | F]¶- default: F
MD_atom_timestep=float¶- default: 0.1 (ASE time units)
MD_atom_timestep_max=float¶- default: 2.0 (ASE time units)
MD_atom_energy_fuzz=float¶- Tolerance for rejecting non-energy conserving trajectories, as fraction of Kinetic Energydefault: 1.0e-2
MD_atom_reject_energy_violation=[ T | F ]¶- Use energy conservation violation (exceeding MD_atom_energy_fuzz * KE) to reject MD trajectories.default: F
python_MD=[ T | F ]¶- Do MD using python code rather than underlying driverdefault: F
atom_velo_rej_free_fully_randomize=[T | F]¶- If true, randomize velocities completely rather than just perturbing.default: F
atom_velo_rej_free_perturb_angle=float¶- Max angle in radians for random rotations.default: 0.3
MC_atom_velo_step_size=float¶- default: 50.0
MC_atom_velo_step_size_max=float¶- default: 10000.0
MC_atom_velo_walk_rej_free=[T | F]¶- default: T. If true, use rejection free algorithm for MC_atom_walk
MC_cell_P=float¶- Pressure value to be used. The unit of pressure depends on both the energy calculator and on the potential model used. Note that ASE useseV as energy and Angstrom as distance units everywhere, thus this case the pressure in the input have to be eV/Angstrom^3 (in case of using LAMMPSthe lammpslib module will convert the units for lammps to whatever units are set by the potential type)default: 0.0
MC_cell_flat_V_prior=[T | F]¶- Flat prior for V (use with MC_cell_P=0 and cell moves, requires reweighting configurations when analyzing)POORLY TESTED, DO NOT TRUST (YET).default: F
MC_cell_volume_per_atom_step_size=float¶- Initial volume stepsize for volume change.default: 5% of the maximum allowed volume
MC_cell_volume_per_atom_step_size_max=float¶- Maximum allowed volume step size.default: 50% of the maximum allowed volume
MC_cell_volume_per_atom_prob=float¶- default: 1.0
MC_cell_stretch_step_size=float¶- default: 0.1
MC_cell_stretch_step_size_max=float¶- default: 1.0
MC_cell_stretch_prob=float¶- default: 1.0
MC_cell_shear_step_size=float¶- default: 0.1, in units of (max_volume_per_atom * N_atoms)^(1/3)
MC_cell_shear_step_size_max=float¶- default: 1.0, in units of (max_volume_per_atom * N_atoms)^(1/3)
MC_cell_shear_prob=float¶- default: 1.0
MC_cell_min_aspect_ratio=float¶- Smallest allowed distance between parallel faces for cell normalised to unit volume. A higher value of
MC_cell_min_aspect_ratiorestricts the system to more cube-like cell shapes, while a low value allows the system to become essentially flat. In case of 64 atoms the use ofMC_cell_min_aspect_ratio< 0.65 does effect the melting transition.default: 0.8 cell_shape_equil_steps=int¶- default: 1000
full_auto_step_sizes=[T | F]¶- If true (T), automatically calibrate all sizes by performing additional short explorations, including at the start of run. If false (F), use initial input step sizes and make small adjustments to these during the run.default: T
monitor_step_interval_times_fraction_killed=float¶- Divided by
n_cull/n_walkersto get actual monitoring interval in iterations, negative for only using last iteration, 0 for no monitoringdefault: 1 adjust_step_interval_times_fraction_killed=float¶- Divided by
n_cull/n_walkersto get actual step adjustment interval in iterations, negative for only using last iteration, 0 for no adjust.default: 1 MC_adjust_step_factor=float¶- default: 1.1
MC_adjust_min_rate=float¶- default: 0.25
MC_adjust_max_rate=float¶- default: 0.75
GMC_adjust_min_rate=float¶- default: 0.25
GMC_adjust_max_rate=float¶- default: 0.75
GMC_dir_perturb_angle=float¶- default: -1.0
GMC_dir_perturb_angle_during=float¶- default: 0.0
MD_adjust_step_factor=float¶- default: 1.1
MD_adjust_min_rate=float¶- default: 0.5
MD_adjust_max_rate=float¶- default: 0.95
ASE_calc_module=str¶- MANDATORY if energy_calculator=ASE
FORTRAN_model=str¶- MANDATORY if energy_calculator=fortran
FORTRAN_model_params=str¶- parameters (optional) for fortran model
LAMMPS_fix_gmc=[T | F]¶- default: F
LAMMPS_init_cmds=str¶- MANDATORY if energy_calculator=lammps
LAMMPS_name=str¶- ‘’, arch name for lammps shared object file
LAMMPS_serial=[T | F]¶- default True, lammps library is serial so do not pass COMM_SELF as a communicator
LAMMPS_header=str¶- lammpslib.py value default, override lammpslib.py header commands for energy_calculator=lammps
LAMMPS_header_extra=str¶- ‘’, extra lammpslib.py header commands for energy_calculator=lammps
LAMMPS_atom_types=str¶- MANDATORY if energy_calculator=lammpsatomic_symbol1 lammps_type1 [, atomic_symbol2 lammps_type2, …]mapping between atom species and lammps potential types
config_file_format=str¶- File format in which configurations are printed, e.g. for trajectory and snapshot files.default: extxyz
rng=( numpy | internal | rngstream )¶- Random number generator.default: numpy
profile=rank_to_profile¶- default: -1
2D=[ T | F ]¶- Perform 2D simulation. Some functionality may be limited with this option!The 2D area is in the xy plane, the z axis stays constant during the simulation (set its value using the keyword
Z_cell_axis).The maximum allowed volume is still defined as volume in this case!If constant pressure is set, the p*V term is now p*A*Z_cell_axis, thus set the MC_cell_P parameter multiplied by 1/Z_cell_axis.default: F Z_cell_axis=float¶- Only used if 2D=T. Value of the Z cell axis, perpendicular to the area in case of a 2D simulation.This stays constant during the simulation.default: 10.0
debug=int¶- Verbosity level used in the output file. The larger its value the more info is printed.default: 0
snapshot_interval=int¶- Iteration interval at which a snapshot is created: every process prints out its current walkers in extended xyz format. If it is set <=0, no snapshots will be printed except the final positions at the end of the nested sampling run. Note that when new snapshots are printed, the previous set is deleted. The snapshot files are convenient source to see how the sampling progresses, but these are also the basis to restart a sampling! When using restart, the walkers will be read from these files.default: 10000
snapshot_time=int¶- Max time between snapshots in secondsdefault: 3600
snapshot_per_parallel_task=[ T | F ]¶- Save a separate snapshot file for each parallel task. Faster I/O (in parallel), but less convenient in some cases.default: T
snapshot_clean=[ T | F ]¶- If true, delete previous iteration snapshot filesdefault: T
random_initialise_pos=[ T | F ]¶- If true, randomize the initial positionsdefault: T
random_initialise_cell=[ T | F ]¶- If true, randomize the initial cell (currently by random walk)default: T
LAMMPS_molecular_info=[ T | F ]¶- If true, create lammps bonds and other molecular MM features from initial atoms config (e.g. can be read from a file)default: T
initial_walk_N_walks=int¶- number of initial random walks to apply to all walkersdefault: ‘’
initial_walk_adjust_interval=int¶- in initial walks, interval between walks for adjusting step sizedefault: ‘’
initial_walk_Emax_offset_per_atom=float¶- offset (per atom) to increase Emax during initial random walk applied to all walkersdefault: 1.0
initial_walk_only=[T | F]¶- do initial walk only, then quitdefault: F
traj_interval=int¶- Iteration interval at which the currently culled configuration(s) is/are printed to the trajectory output, in the set format. If it is set <=0, no trajectory files will be printed at all. Useful option for larger runs as the trajectory files can become huge.default: 100
delta_random_seed=int¶- Random number seed to be used in the run. If smaller than 0, a seed from /dev/urandom is used.default: -1
no_extra_walks_at_all=[ T | F ]¶- default: F
track_configs=[ T | F ]¶- Track configrations across all walks/clones.default: F
track_configs_write=[ T | F ]¶- Write tracked configrations to a file (if false, tracking info will still be in snapshot and traj files)default: F
ns_run_analyzers=string¶- Analyzers to apply during run. String consists of semicolon separated pairs of module name and intervals. Module names correspond to analysis modules in NS_PATH/ns_run_analyzers (see there for examples) or elsewhere in PYTHONPATHPositive interval refers to NS loop, negative to initial walksdefault:’’
min_nn_dis=float¶- Optional input to add additional rejection criteria when initially generating walkers. If any nearest neighbour distances are less than this length, given in Angstroms, the walker is rejected.default: 0.0
calc_nn_dis=[ T | F ]¶- While you can set the min_nn_dis to add an additional rejection criteria to the initial walker generation, you can use this keyword to keep the rejection criteria on throughout the sampling. This does increase the calculation time of a run and scales exponentially with the number of atoms.default: F
Example input files¶
LJ Cluster with the supplied fortran code, using MC¶
This input file will start a calculation of 6 Lennard-Jones atoms in a cubic cell with volume 648 Angstrom^3, using the potential implemented in the supplied fortran routine, and using MC trajectory for generating a new sample configuration. As the shape and volume of the cell will not be changed, a cluster will be formed by the atoms. Note that the input parameters given below, such as the number of walkers and length of trajectory, are for a short test run. For six atoms this calculation should take only a couple of minutes on one processor, and although the calculation should end up in the global minimum octahedral cluster structure, thermodynamic variables will not be necessarily well converged.
# starting volume (in this example it is Angstrom^3)
max_volume_per_atom=108.0
# 6 atoms with atomic number 1 and mass 1.0
start_species=1 6 1.0
start_energy_ceiling_per_atom=0.17e9
# size of live set
n_walkers=128
# at each iteration 1 walker is killed (n_cull=1 is strongly recommended)
n_cull=1
# total number of iterations will be n_iter_times_fraction_killed /(n_cull/n_walkers)
n_iter_times_fraction_killed=100
# prefix of all the output files
out_file_prefix=test.cluster.MC.fortran
# use MC generating a new walker
atom_algorithm=MC
n_model_calls_expected=3
n_atom_steps=5
# length of atomic trajectory in each step
atom_traj_len=5
# number of cell volume steps (zero as the cell should not change for the cluster)
n_cell_volume_steps=0
# number of cell shear steps (zero as the cell should not change for the cluster)
n_cell_shear_steps=0
# number of cell strectch steps (zero as the cell should not change for the cluster)
n_cell_stretch_steps=0
FORTRAN_model=example_LJ_min_image_model.so
# use the supplied fortran code
energy_calculator=fortran
rng=numpy
# verbosity level
debug=0
Periodic LJ with LAMMPS, using MD¶
This input file will start a calculation of 64 Lennard-Jones atoms
in a cell with variable shape and size at the set pressure value, using the potential implemented in LAMMPS
and using MD trajectory for generating a new sample configuration.
# starting volume (in this example it is Angstrom^3)
max_volume_per_atom=1000000.0
# 64 atoms with atomic number 1, let mass default to 1.0
start_species=1 64
start_energy_ceiling_per_atom=1.5
# size of live set
n_walkers=256
# at each iteration 1 walker is killed
n_cull=1
# total number of iterations will be n_iter_times_fraction_killed / (n_cull/n_walkers)
n_iter_times_fraction_killed=5000
# prefix of all the output files
out_file_prefix=test.periodic.MD.lammps
# use MD generating a new walker
atom_algorithm=MD
n_model_calls_expected=8
n_atom_steps=5
# length of atomic trajectory in each step
atom_traj_len=5
# number of cell volume steps
n_cell_volume_steps=3
# number of cell shear steps
n_cell_shear_steps=2
# number of cell strectch steps
n_cell_stretch_steps=2
# Pressure (in the units of the potential)
MC_cell_P=2.37e-2
# allowed torsion for the cell, ratio of the min lattice height
MC_cell_min_aspect_ratio=0.9
# use LAMMPS
energy_calculator=lammps
# name of the LAMMPS machine (the one that *YOUR* LAMMPS was built with)
LAMMPS_name=mpi
# potential parameters
LAMMPS_init_cmds=pair_style lj/cut 9.00; pair_coeff * * 0.1 3.0; pair_modify shift yes
LAMMPS_atom_types=H 1
rng=numpy
# verbosity level
debug=0
Periodic binary LJ with the supplied fortran code or LAMMPS, using MD¶
This input file will start a calculation of a binary Lennard-Jones system, with 32 A-type atoms and 32 B-type atoms. Note that in case of multicomponent systems swap moves have to be introduced, when the coordinates of different atomic types are swapped. The cell is of variable shape and size at the set pressure value.
# starting volume, in A^3
max_volume_per_atom=2000.0
# 64 atoms total, 32 of each type
# masses will default to 1.0, which is efficient and correct
start_species=1 32, 2 32
# prevent atoms completely on top of each other
start_energy_ceiling_per_atom=1.5
n_walkers=9216
n_cull=1
# total number of iters will be 1500/(1/9216) = 1500*9216
n_iter_times_fraction_killed=1500
# could also stop iteration when convergence is assured for T > some cutoff
# converge_down_to_T=200
# or could stop convergence once energy is low enough
# min_Emax=-61.0
out_file_prefix=test.periodic_binary.MD.FORTRAN_LJ
################################################################################
atom_algorithm=MD
n_model_calls_expected=512
n_atom_steps=1
atom_traj_len=8
n_cell_volume_steps=32
n_cell_shear_steps=8
n_cell_stretch_steps=8
n_swap_steps=8
################################################################################
# pressure in units of the potential (eV/A^3), this is 0.1 GPa
MC_cell_P=6.25e-4
# default MD time steps and adjustment rates should be correct
# default cell steps should be correct
MC_cell_min_aspect_ratio=0.85
################################################################################
# binary LJ from hardcoded FORTRAN
FORTRAN_model=example_LJ_model.so
energy_calculator=fortran
# LAMMPS is another possibility
# energy_calculator=lammps
# name of the LAMMPS machine (i.e. the part of the library file that follows 'liblammps_', without the '.so' suffix)
# LAMMPS_name=mpi
# # potential parameters
# LAMMPS_init_cmds=pair_style lj/cut 9.00; pair_coeff 1 1 0.1 3.0; pair_coeff 1 2 0.2 3.0; pair_coeff 2 2 0.1 3.0; pair_modify shift yes
# conversion from atomic numbers (set in start_species) to types for LAMMPS
# LAMMPS_atom_types=H 1,He 2
rng=numpy
debug=0
Molecule with LAMMPS, using MD¶
Start a nested sampling simulation with a polymer in a cell with constant volume and shape. Initially the random placement of atoms has to be turned off,
and a configuration file has to be read. The initial walk is done with a heuristic choice for E_max.
The atom_style full used in this example is part of the MOLECULE package, you have to include that when compiling
LAMMPS.
# starting volume (in this example it is Angstrom^3)
max_volume_per_atom=108.0
# read in the initial configuration of the polymer
start_config_file=test_start_config_polymer.xyz
start_energy_ceiling_per_atom=0.17e9
# switch off initial random displacement
random_initialise_pos=F
random_initialise_cell=F
initial_walk_N_walks=20
#initial_walk_adjust_interval=10
initial_walk_Emax_offset_per_atom=1.0
# size of live set
n_walkers=128
# at each iteration 1 walker is killed (n_cull=1 is strongly recommended)
n_cull=1
# total number of iterations will be n_iter_times_fraction_killed / (n_cull/n_walkers)
n_iter_times_fraction_killed=100
# prefix of all the output files
out_file_prefix=test.cluster.MD.lammps.polymer
# use MD generating a new walker
atom_algorithm=MD
n_model_calls_expected=3
n_atom_steps=5
# length of atomic trajectory in each step
atom_traj_len=5
# number of cell volume steps (zero as the cell should not change for the cluster)
n_cell_volume_steps=0
# number of cell shear steps (zero as the cell should not change for the cluster)
n_cell_shear_steps=0
# number of cell strectch steps (zero as the cell should not change for the cluster)
n_cell_stretch_steps=0
energy_calculator=lammps
# name of the LAMMPS machine (the one that *YOUR* LAMMPS was built with)
LAMMPS_name=mpi
# potential parameters
LAMMPS_init_cmds=pair_style lj/cut 1.50; pair_coeff * * 1.0 1.0; pair_modify shift yes; bond_style harmonic; bond_coeff 1 30.0 1.5; special_bonds lj 0.0 0.0 1.0; angle_style harmonic; angle_coeff 1 30.0 180.0
LAMMPS_header_extra=atom_style full
LAMMPS_atom_types=H 1
rng=numpy
# verbosity level
debug=5
The corresponding test_start_config_polymer.xyz file is the following: (note that _ is an underscore not a -)
6
Lattice="10.0 0.0 0.0 0.0 10.0 0.0 0.0 0.0 10.0" Properties=species:S:1:pos:R:3:bonds:S:1:angles:S:1
H 0.0 0.0 0.0 1(1) _
H 1.5 0.1 0.0 2(1) 0-2(1)
H 3.0 0.0 0.0 _ _
H 0.0 0.0 5.0 4(1) _
H 1.5 0.1 5.0 5(1) 3-5(1)
H 3.0 0.0 5.0 _ _
If multiple bonds or angles have to be defined per atom, use a comma to separate the entries without spaces, and if no entries are needed for an atom still use a single underscore.
Some tips on setting the input parameters¶
Setting the random walk parameters¶
When the walker configuration with the highest energy is chosen and replaced by a clone of
another randomly picked walker, this clone has to be perturbed in order to satisfy the criteria
that all walkers should be uniformly distributed in the available phase space. This perturbation
is done by performing a random walk, modifying the atomic coordinates and the lattice parameters
if applicable. This can be done by one of the following different methods: MC, MD or GMC,
set by the atom_algorithm keyword. Although infinitely long walks would be the best,
we can rarely wait that long, thus we have to determine a finite length we allow for the walk.
The length is determined by “step”, i.e. the number of model calls. One MD timestep, one lattice
modification, one atom type swap or one MC all-atom sweep is counted as a single model call.
The number of model calls a user wants to be performed on a walker between its birth (by cloning) and
discard (having the highest energy among all the walkers) is set by the n_model_calls_expected
keyword. If the sampling is run in parallel, the code performs approximately n_model_calls_expected * n_cull / number_of_processors
model calls on each processors at each iteration. This means that all walkers will perform
n_model_calls_expected steps on average. Note that the code will continue to do
steps until it has done at least the requested number of calls, so this number may sometimes
be exceeded, e.g. if the last step is a long one (e.g. an atomic trajectory with multiple steps).
Currently the following step types are allowed: changing atomic coordinates, changing the volume of the
cell, changing the shape of the cell by shear, changing the shape of the cell by stretch, and swapping
coordinates of different types of atoms. The ratio of these steps are determined by the keywords
n_atom_steps, n_cell_volume_steps, n_cell_shear_steps, n_cell_stretch_steps and n_swap_steps,
respectively. E.g. if the values for these keywords are set as 10, 4, 4, 4, 3, respectively, then the probability
of performing an atomic step will be 10/(10+4+4+4+3)=0.4, the probability of performing a volume change will be 0.16,…etc.
If any of these keywords is set to zero, the corresponding step type will never be performed.
It is important to consider that one n_atom_steps does not necessarily cover only one model call. To improve efficiency
several MD time steps or MC sweeps can be (and should be) performed consecutively, set by the parameter atom_traj_len.
If atom_traj_len=8 and an atomic step is chosen, then 8 model calls will be performed out of the
total n_model_calls_expected.
Minimum lattice height: MC_cell_min_aspect_ratio¶
The fully flexible cell is introduced to remove the finite size effect whereby it may not be possible to arrange a fixed number of particles in a fixed shape cell into certain crystal structures. In unfortunate cases this can exclude thermodynamically relevant structures from the results of the calculation. But for a flexible cell and for a finite number of particles, there exist simulation cells such that parallel faces of the cell are separated by only a few layers of atoms, and those does not approximate the infinite fluid in three dimensions. This problem can be solved by the introduction of a “minimum cell height” parameter.
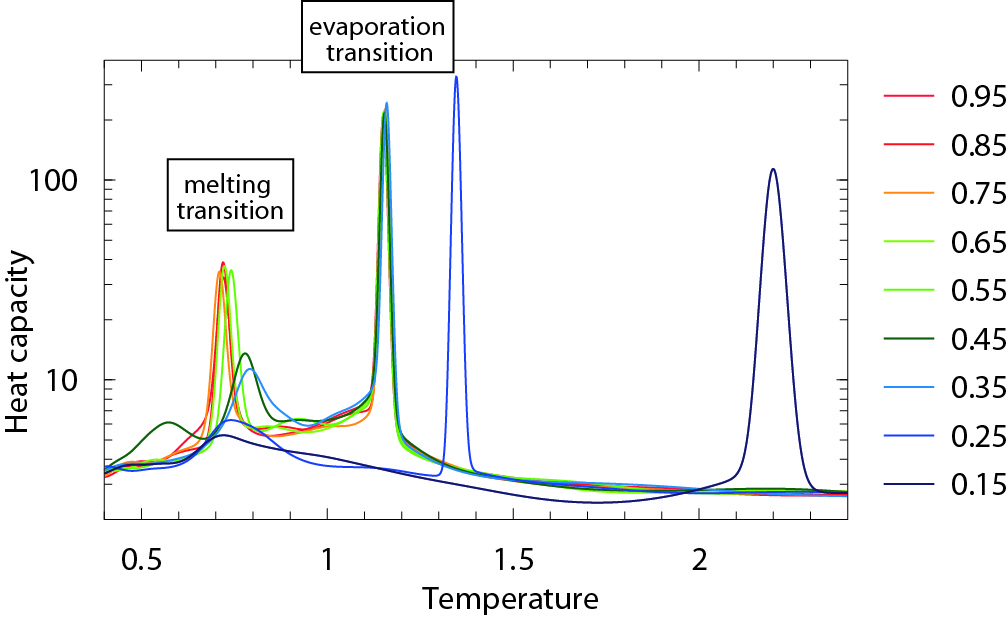
The effect of the chosen minimum cell height is demonstrated in the case of the periodic system of
64 Lennard-Jonesium particles in the figure below. The legend on the right shows the value of
MC_cell_min_aspect_ratio used in each simulation. The peak at lower temperature corresponds to melting,
and the peak at higher temperature to evaporation. The location of
the evaporation transition is converged for MC_cell_min_aspect_ratio ≥ 0.35, but melting requires
a higher MC_cell_min_aspect_ratio ≥ 0.65. At low values of MC_cell_min_aspect_ratio the
system’s behaviour is dominated by the fictitious periodicity imposed by the boundary
conditions.
(source: R. J. N. Baldock, Classical Statistical Mechanics with Nested Sampling, Ph.D. thesis, University of Cambridge (2014).)
Restart a run¶
If one needs to continue a run, i.e. perform more iteration cycles, the input file has to be modified with adding the following line
restart_file=my_output.snapshot.all.extxyz
Include the keyword restart_file. This defines the name of the file
where all the walker configurations can be read from. This file should be produced by
concatenating the last saved snapshot files of all the processors. You might also need to increase the n_iter_per_walker
value if the run already reached the number of iterations set previously. (Note: you do not have to comment out the start_species option.)